Maladie de Huntington : Que peuvent apporter les cellules souches ?
La maladie de Huntington (HD) est une maladie neurodégénérative héréditaire aux effets dévastateurs qui touche environ 1 personne sur 10 000 aux Etats Unis, en Europe et en Australie. Elle provoque habituellement des mouvements irrépressibles du visage et du corps, et la démence. Les symptômes empirent au cours du temps, la personne atteinte devient totalement dépendante de l'aide des autres. A l’heure actuelle, il n’existe aucun traitement efficace. Comment la recherche sur les cellules souches pourrait-elle conduire à de nouveaux traitements ?
Que savons nous ? ▼
La maladie de Huntington (HD) est une maladie génétique héréditaire qui détruit les neurones épineux moyens (MSN) du cerveau, provoquant des modifications de l’humeur, un déclin des facultés mentales et des problèmes de motricité.
Les symptômes apparaissent souvent chez des patients entre 35 et 50 ans, mais peuvent commencer plus tôt.
La HD est due à la répétition plus de 40 fois de la séquence d’ADN ‘CAG’ dans le gène de la Huntingtine, les personnes avec plus de 35 répétitions étant à risque de développer la maladie.
A l’heure actuelle, il n’existe aucun traitement de la maladie de Huntington.
Les cellules souches ont été très utiles dans l’étude de nombreux aspects de la maladie, depuis la façon dont la maladie provoque la mort des MSN jusqu’aux essais de nouveaux médicaments.
Sur quoi travaillent les chercheurs ? ▼
Le gène de la Huntingtine code pour une protéine essentielle pour les neurones, mais les scientifiques essaient encore de comprendre le rôle de cette protéine dans les cellules et les raisons pour lesquelles un trop grand nombre de répétitions ‘CAG’ entraîne la mort des MSN.
Les chercheurs utilisent des MSN produits avec des cellules souches pluripotentes induites (cellules iPS) pour modéliser la HD et observer la façon dont elle progresse. Les cellules iPS sont aussi utilisées pour tester rapidement de nouveaux médicaments, des thérapies géniques et d’autres traitements en moins de temps qu’avant de disposer des cellules iPS.
Des études portent sur la façon dont on pourrait transplanter des cellules souches du cerveau (neurales) dans des cerveaux de patients pour le réparer et éventuellement régénérer des MSN et d’autres cellules cérébrales nécessaires.
Quels sont les défis ? ▼
Aucun traitement de la HD par cellules souches n’est actuellement approuvé. Plusieurs essais de greffes de cellules souches neurales ont été réalisés pour traiter des patients atteints de HD, mais ces traitements doivent encore faire l’objet d’essais cliniques rigoureux pour démontrer leur innocuité et leur efficacité.
Il faut développer des sources de cellules souches neurales qui soient éthiques et fiables. Il serait possible de produire de grandes quantités de cellules souches neurales avec des cellules iPS ou des cellules souches embryonnaires, mais ces cellules doivent être homogènes, stables et sûres. Si elles ne sont pas correctement testées, les cellules souches pluripotentes pourraient être à l’origine de cancers ou d’autres complications mortelles.
- Qu’est ce que la maladie de Huntington ?
- HD et ‘répétitions CAG’
- Quels sont les traitements actuels de la Maladie de Huntington ?
- Quelle peut être la contribution des cellules souches ?
- Recherche actuelle
- Peut-on actuellement utiliser les cellules souches pour traiter la HD?
- En savoir plus
- Find out more (English)
- Remerciements et références
La maladie de Huntington (HD) atteint essentiellement les cellules nerveuses du cerveau appelées neurones épineux moyens (MSN). Les MSN reçoivent et coordonnent les informations provenant d’autres neurones du cerveau pour contrôler les mouvements du corps, du visage et des yeux.
Dans la maladie de Huntington, un grand nombre de MSN sont endommagés ou détruits. Il semble que d’autres types de neurones du cerveau soient aussi affectés, comme les neurones corticaux. Les patients notent habituellement les premiers symptômes lorsqu’ils ont entre 35 et 50 ans, généralement de faibles mouvements spasmodiques des muscles du visage et des membres. Au fur et à mesure de l’évolution de la maladie, ces mouvements spasmodiques deviennent plus évidents et fréquents et d’autres symptômes apparaissent. Les patients sont affectés de différentes façons, mais les symptômes peuvent comprendre des difficultés d’élocution et de déglutition, une démence ou des problèmes de concentration
HD est une maladie héréditaire. Les enfants dont un parent est atteint ont un risque de 50% d’hériter de l’anomalie génétique responsable de la maladie. Cette anomalie se produit dans le gène qui code pour une protéine appelée Huntingtine. Le gène défectueux entraine la production par l’organisme d’une version erronée et toxique de la protéine Huntingtine, ce qui se traduit finalement par la perte de MSN et d’autres neurones.
L’alphabet de l’ADN est composé de quatre lettres : C, A, G et T. Ce sont les quatre acides aminés qui sont associés dans des ordres différents pour composer les brins d’ADN. Dans sa version non pathologique (sans maladie), le gène de la Huntingtine comporte jusqu’à 35 répétitions de la séquence CAG et, sous cette forme, le gène est bénéfique aux neurones.
Néanmoins, les scientifiques ont découvert que le gène est d’abord apparu sans aucune répétition CAG, dans une amibe appelée Dictyostelium discoideum. Les CAG sont apparus et ont augmenté en nombre au fur et à mesure que les espèces se développaient avec des systèmes nerveux de plus en plus complexes.
Il semble qu’il pourrait y avoir une corrélation entre le nombre de répétitions CAG dans le gène normal et les capacités neuronales et que, chez les êtres humains en bonne santé, plus le gène contiendrait de CAG (jusqu’à un maximum de 35), plus il y aurait de matière grise dans le cerveau. Il se pourrait ainsi qu’il y ait corrélation entre les répétitions CAG et la complexité et la fonction cérébrales aussi bien au cours de l’évolution que chez les êtres humains, bien que ceci n’est pas encore été prouvé et reste simplement, pour le moment, une hypothèse intéressante.
Ce qui est clair est qu’un nombre de répétitions CAG supérieur à 35 provoque la maladie de Huntington chez les êtres humains.
Actuellement il n’y a pas de traitements efficaces pour stopper ou inverser l’évolution de la maladie. Les traitements sont plutôt focalisés sur la prise en charge des symptômes. Les médicaments les plus couramment utilisés pour traiter les mouvements incontrôlés sont la tetrabenazine, les benzodiazepines et les antipsychotiques, ces derniers pouvant aussi soulager quelques uns des possibles troubles psychologiques. L’orthophonie est largement utilisée pour améliorer la communication et les difficultés à s’alimenter et à déglutir (avaler). D’autres médicaments peuvent aussi être utilisés en fonction des besoins et symptômes de chaque patient.
Ces traitements lourds ont souvent des effets secondaires et ne remédient pas aux dommages ni ne stoppent la progression de la maladie. C’est pourquoi les scientifiques font des efforts considérables pour trouver de nouvelles méthodes pour s’attaquer à la cause de la maladie et pour remplacer les neurones épineux moyens perdus.
Bien que les chercheurs sachent que la HD est provoquée par une erreur dans le gène qui fabrique la protéine Huntingtine, la façon dont l’erreur génétique conduit à la perte des neurones épineux moyens et aux symptômes qui en résultent n’est pas claire. La compréhension de ce processus pourrait conduire à de nouvelles options thérapeutiques. Les scientifiques espèrent aussi trouver des moyens de remplacer les MSN détruits chez les patients.
Les cellules souches offrent l’opportunité de cultiver et d’étudier en laboratoire de grands nombres de cellules. Il existe plusieurs types de cellules souches et les scientifiques cherchent les différents moyens qu’ils pourraient utiliser pour relever les défis de la maladie de Huntington :
- Etudier et comprendre la maladie : Les chercheurs peuvent utiliser certaines sortes de cellules souches pour cultiver de grandes quantités de types de cellules endommagées par la HD. Ces cellules peuvent être utilisées en recherche pour découvrir exactement le fonctionnement de la maladie et quelle(s) est (sont) la (les) fonction(s) du gène normal dans un cerveau sain.
- Remplacer les cellules perdues : dans le futur, les scientifiques espèrent pouvoir utiliser les cellules souches pour cultiver de nouveaux MSN sains qui pourront être transplantés chez les patients en remplacement des cellules détruites par la maladie.
- Développer de nouveaux médicaments : Certains types de cellules souches pourraient être utilisés pour cultiver des MSN porteurs de l’anomalie génétique responsable de la maladie de Huntington. Ces cellules pourraient être un outil précieux pour rechercher et tester de nouveaux médicaments.
Le remplacement des cellules perdues
La majorité (mais pas tous) des symptômes de la HD sont dus à la perte des neurones épineux moyens du cerveau. Par conséquent les efforts des scientifiques se sont focalisés sur l’obtention de nouveaux MSN pour remplacer ceux endomagés. En 2000, des médecins ont transplanté des neurones fœtaux dans le cerveau d’un petit nombre de patients atteints de HD. Ces patients ont connu une amélioration de courte durée des symptômes moteurs et psychologiques, mais deux ans après l’intervention chirurgicale les symptômes se sont à nouveau aggravés. Il est possible que l’amélioration des procédés d’utilisation de ces MSN fœtaux puissent aider à traiter la HD et des essais cliniques testent actuellement cette hypothèse, mais il y a certaines difficultés techniques et scientifiques importantes : par exemple, l’utilisation de tissus prélevés chez des fœtus issus d’avortements ne fournit qu’une source limitée de cellules qui ne peuvent être ni purifiées ni corrigées. Les cellules souches pourraient fournir une précieuse alternative
Les chercheurs espèrent utiliser les cellules souches embryonnaires et les cellules souches pluripotentes induites comme source illimitée de neurones épineux moyens pour traiter la HD. Ces deux types de cellules souches ont la capacité de fabriquer toutes les cellules de l’organisme, et le défi est donc de trouver un moyen de les conduire à ne fabriquer que des MSN. Les premiers neurones cultivés en laboratoire ressemblant aux MSN avaient été obtenus à partir de cellules souches embryonnaires humaines en 2008, mais ils provoquèrent la croissance de tumeurs lorsqu’ils furent transplantés chez des rongeurs. Plus récemment, en 2012, deux laboratoires ont obtenu des MSN à partir de cellules souches embryonnaires humaines de manière à ce qu’il n’y ait pas formation de tumeurs. La transplantation en laboratoire de tels MSN permet d’obtenir une certaine amélioration de la motricité chez des rongeurs atteints d’une forme de HD. Il a aussi été montré, dans un cas, que les neurones possédaient un certain nombre des propriétés clés attendues chez les véritables MSN du cerveau humain. Il est nécessaire de poursuivre les recherches pour améliorer la qualité et la quantité de neurones obtenus et déterminer si cette approche pourrait être un jour sans danger et transposée efficacement aux patients.
Un certain nombre d’études récentes ont montré qu’il était aussi possible d’obtenir des cellules souches pluripotentes induites (cellules iPS) provenant de patients atteints de maladie de Huntington. Les chercheurs d’un consortium de six laboratoires américains et deux laboratoires français spécialisés dans la HD, ont prélevé des cellules cutanées chez des patients atteints de HD et les ont reprogrammées pour obtenir des cellules iPS. Cela a été également réalisé de manière indépendante par un laboratoire en Californie. Les cellules iPS ainsi obtenues ont le même code génétique que les patients et, de ce fait, les cellules ont aussi le défaut génétique responsable de la HD et montrent certaines des caractéristiques spécifiques à la maladie. Les chercheurs ont corrigé le défaut génétique en laboratoire de sorte que les cellules produisent une protéine Huntingtine pleinement fonctionnelle. Les MSN cultivés à partir de ces cellules iPS génétiquement corrigées semblent être sains : en laboratoire, ils sont aussi résistants à la mort cellulaire que les MSN de personnes sans HD. Beaucoup plus de travaux de recherche sont nécessaires, mais cette approche pourrait devenir un jour un outil important à utiliser dans les traitements de substitution de la HD
Le criblage et la découverte de médicaments
Grâce aux technologies modernes, les chercheurs ont accès à des systèmes automatisés qui leur permettent de tester en un temps relativement court des millions de composés chimiques pour leur possible action bénéfique sur les cellules. Ces systèmes sont des outils précieux pour la découverte de médicaments, mais il y a un problème : ils nécessitent d’importantes quantités du bon type de cellules (des neurones par exemple) et les cellules doivent exprimer la maladie étudiée. Il est impossible d’obtenir directement des patients les MSN avec la maladie de Huntington car ils doivent être chirurgicalement prélevés dans le cerveau. Les cellules iPS engendrées à partir du muscle ou de la peau de patients atteints de HD pourraient résoudre ce problème en fournissant une source illimitée de MSN avec la maladie de Huntington. De nombreuses recherches sont consacrées au développement de systèmes puissants de production de cellules iPS avec la maladie de Huntington pour une telle recherche de nouveaux médicaments.
L’étude du comportement de la maladie
Des mutations (ou changements) dans le gène qui code pour la protéine Huntingtine furent associées pour la première fois à la maladie de Huntington en 1993. Le gène de la Huntingtine contient un fragment d’ADN appelé ‘répétition CAG’ – un morceau d’ADN fait de trois unités (C, A et G) qui apparaissent de nombreuses fois dans le même ordre. Le gène normal, sain, contient moins de 35 répétitions CAG, mais les patients atteints de HD ont une version mutée qui contient plus de 35 répétitions CAG et, dans de très rares cas, jusqu’à 250 répétitions CAG. Ces copies supplémentaires de la séquence CAG produisent une version erronée et toxique de la Huntingtine. Plus le nombre de répétitions CAG est élevé, plus la Huntingtine est toxique, et plus les symptômes tendent à se manifester précocement.
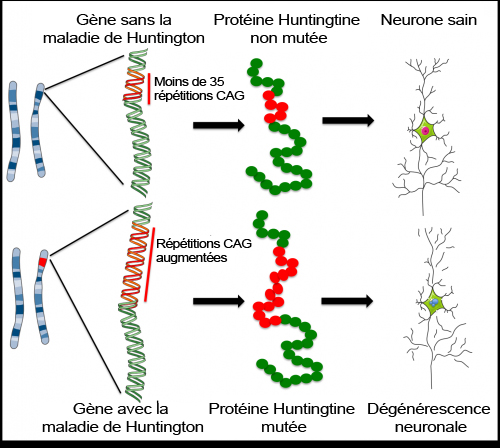
Les scientifiques qui travaillent sur la protéine Huntingtine mutante se concentrent sur l’identification des mécanismes selon lesquels elle contribue à la maladie. Comment la modification dans cette seule protéine endommage-t-elle les MSN ? Mais la Huntingtine interagit avec de nombreuses autres protéines et influe sur de nombreux processus dans nos cellules, et l’on ne connaît toujours pas la façon exacte dont elle contribue à l’évolution de la maladie. Pour étudier cette question, les scientifiques utilisent des modèles de la maladie : des systèmes conçus pour reproduire la HD en laboratoire afin de l’étudier à l’extérieur de l’organisme du patient. Les modèles actuels se basent sur les rongeurs ou les cellules génétiquement modifiées, mais il n’y a pas de modèle parfait qui mime exactement la maladie, et aucun des modèles ne reproduit le lien observé chez les patients entre le nombre de répétitions CAG et la toxicité de la protéine
Les cellules souches pourraient être utilisées pour développer un modèle d’étude plus exact des mécanismes et de la progression de la HD. Les cellules iPS proviennent de patients avec des nombres de répétitions CAG différents. Quand ces différentes cellules iPS sont utilisées pour obtenir des MSN, les neurones qui ont un plus grand nombre de répétitions CAG sont plus vulnérables à différents stress et présentent de nombreuses caractéristiques de la maladie telle qu’elle se manifeste chez les patients. Les chercheurs espèrent que ces systèmes de cellules iPS vont les aider à découvrir quel est le fonctionnement exact de la HD.
L’utilisation des cellules souches pour la recherche sur la HD s’est développée très récemment. Bien que les cellules souches commencent déjà à fournir des outils précieux pour étudier l’évolution de la HD et pour chercher de nouveaux médicaments, il y a encore un long chemin à parcourir avant que l’on puisse considérer la transplantation de cellules chez les patients comme un approche thérapeutique fiable. Il faudra auparavant répondre aux principales questions suivantes
- Quel est le degré de similarité entre les MSN cultivés en laboratoire et ceux trouvés dans nos cerveaux ? Bien que l’on ait montré qu’ils partagent certaines caractéristiques, ils ne sont vraisemblablement pas identiques et doivent être mieux compris afin que l’on puisse développer des méthodes d’évaluation et de maintien de la qualité des cellules cultivées en laboratoire.
- Combien de temps les MSN produits en laboratoire peuvent ils survivre une fois transplantés dans le cerveau et sont ils capables d’y remplir leur rôle?
- Ces MSN produits en laboratoire seront ils capables de s’intégrer dans le système nerveux adulte endommagé, de reconnecter les circuits détruits et d’offrir un bénéfice aux patients?
- L’Implantation de MSN sera-t-elle sans risque pour les patients, particulièrement dans le long terme?
C’est pourquoi il n’est pas possible aujourd’hui d’utiliser les cellules souches pour traiter la HD , mais la recherche sur ces cellules fournit des outils utiles aux scientifiques en vue d’élaborer de nouvelles approches pour l’avenir.
Cette fiche d’information a été élaborée par Serafi Cambray et révisée par Elena Cattaneo.
Image principale des neurones cultivés a partir de cellules souches embryonnaires par Serafi Cambray. Neurones épineux moyens labelled with green fluorescent proteins par Valentina Castiglioni. Neurones épineux moyens humains cultivés a partir de cellules souches embryonnaires par Charles Arber. Toutes les autres images et schémas par Serafi Cambray. Schéma créé avec Servier Medical Art.
Dernière mise à jour: