Huntington’s disease: how could stem cells help?
Huntington’s disease is a devastating, hereditary neurodegenerative disease affecting about 5 out of every 100,000 people in the USA, Europe and Australia. It typically causes involuntary movements of the face and body, and dementia. Symptoms worsen over time, eventually leaving the affected person totally dependent on help from others. There are no effective therapies available today. How might stem cell research lead to new treatments?
What do we know? ▼
Huntington’s disease (HD) is a genetically heritable disease that destroys ‘medium spiny neurons’ (MSNs) in the brain, leading to emotional changes, mental decline and impaired movement.
Symptoms often appear in patients between the ages of 35-50, but can start earlier.
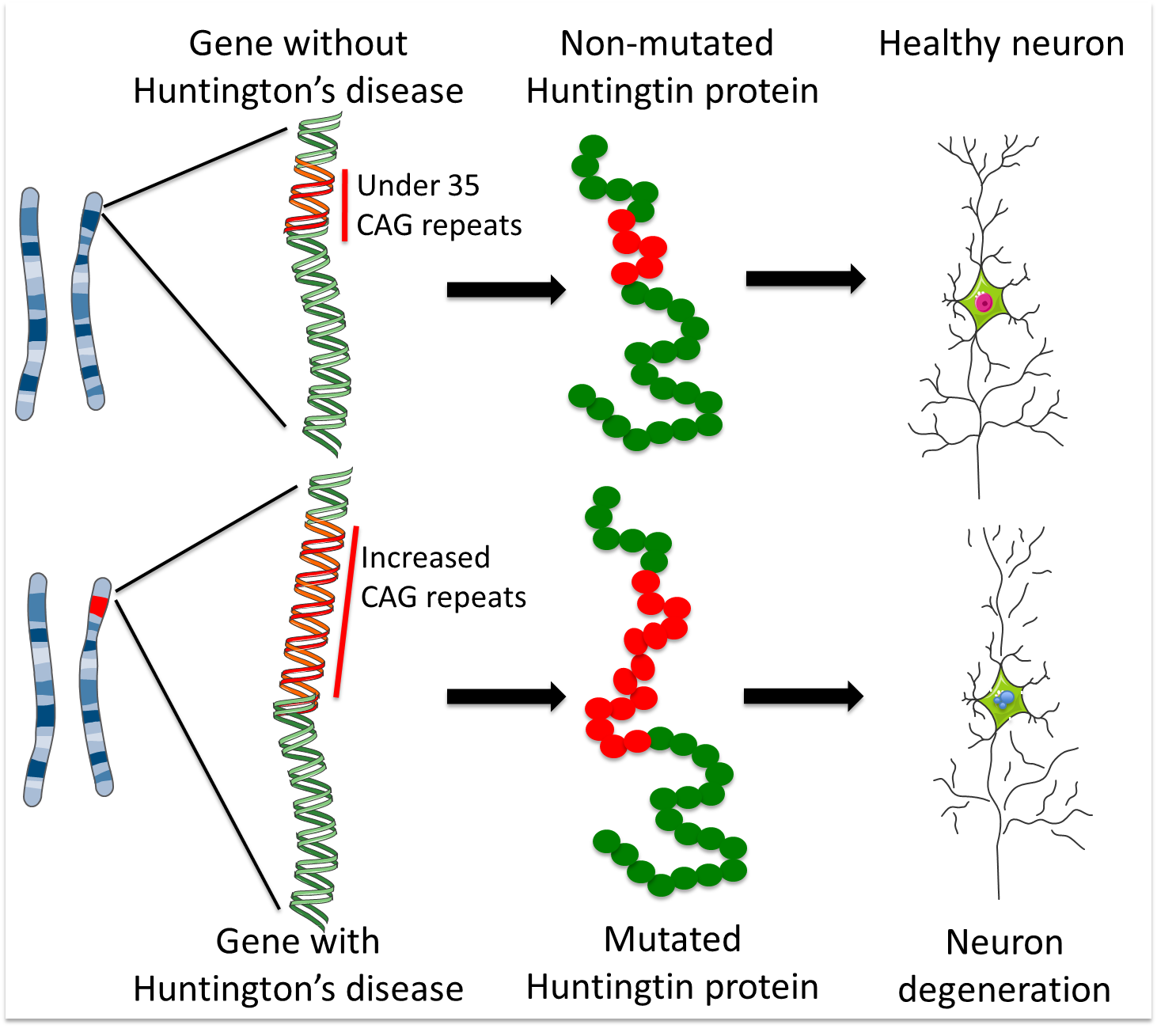
HD is caused by a ‘CAG’ sequence of DNA being repeated >40 times in the Huntingtin gene, with people with >35 repeats at risk of developing HD.
There are currently no treatments for Huntington’s disease.
Stem cells have been an invaluable tool for studying many aspects of HD, from how HD causes MSNs to die to testing new treatments.
What are researchers investigating? ▼
The Huntingtin gene makes a protein important for neurons, but scientists are still trying to understand what this protein does in cells and why too many ‘CAG’ repeats causes MSNs to die.
Researchers are using MSNs made with induced pluripotent stem cells (iPSCs) to model HD and observe how HD progresses. iPSCs are also used to rapidly test new HD drugs, gene therapies and other treatments faster than before iPSCs were available.
Studies are examining how brain (neural) stem cells could be transplanted in patients’ brains to repair the brain and possibly regrow MSNs and other needed brain cells.
What are the challenges? ▼
Currently there are no approved stem cell treatments for HD. Neural stem cell transplants have undergone several trial studies for treating HD patients, but these treatments must still undergo rigorous clinical trials to show they are safe and effective.
Ethical and reliable sources of neural stem cells must be developed. iPSCs and embryonic stem cells may be able to make large numbers of neural stem cells, but these cells must be uniform, predictable and safe. If they are not properly tested pluripotent stem cells could cause cancer or other life-threatening complications.
- About Huntington's disease
- Huntington’s Disease and 'CAG repeats'
- How is Huntington’s disease treated now?
- How could stem cells help?
- Current research
- Can stem cells be used to treat HD now?
- Find out more
- Acknowledgements and references
Huntington’s Disease (HD) mainly affects nerve cells in the brain called medium spiny neurons (MSNs). MSNs receive and coordinate information from other neurons in the brain to control movement, intellectual processes and emotion. Although the most well known movement abnormalities in Huntington’s are the involuntary jerky movements called chorea, Huntington’s causes a range of movement abnormalities including slowing of movements, dystonia (more prolonged and twisting movements than seen with chorea), difficulty coordinating accurate movements and abnormalities of balance. The movement abnormalities can affect widespread areas of the body, including those of the trunk limbs, face, eyes, and the muscles controlling speech and swallowing. The movement abnormalities are the most obvious sign of the condition, but the disturbance of intellectual processes and emotion often cause the most disability.
In HD large numbers of MSNs are damaged and destroyed. Some other types of neurons in the brain also appear to be affected, such as cortical neurons. The most common age for people developing movement symptoms is when they are around 35-50 years old, although the symptoms can start much earlier or later than that, and some of the emotional and thinking problems can occur in a subtle way long before the movement problems arise. People can be affected in different ways, especially at in the early years of the condition, but in all the condition will progress over a period of two to three decades.
HD is an hereditary disease. Children with an affected parent have a 50% chance of inheriting the genetic fault that causes the disease. This fault occurs in the gene that holds the code for a protein called Huntingtin. The defective gene causes the body to make a faulty, toxic version of the Huntingtin protein and this eventually results in the loss of MSNs and other neurons.
The 'DNA alphabet' is made up of four letters: C, A, G and T. These are the four nucleic acid units that are combined in different orders to make DNA strands. In its non-disease version, the Huntingtin gene contains up to 35 repeats of the sequence CAG. More than 40 repeats result in a person developing Huntington’s disease at some stage in their life (although the age at which they will develop it remains very difficult to predict). 36-39 repeats is a grey area; individuals with these repeat numbers may develop Huntington’s (the risk increases as the repeat number increases), but some may not develop it within their life-time.
Interestingly, scientists discovered that the gene first appeared without any CAG repeats at all, in an amoeba called Dictyostelium discoideum. As new species evolved, the CAGs in the huntingtin gene appeared and increased in number in species with progressively more complex nervous systems, suggesting that the longer CAG repeats confer a beneficial effect on neurons.
There is currently no effective treatment to stop or reverse the progress of HD, but there are some ways of helping to make the symptoms more manageable. For example, some drugs are available which may help to dampen down the involuntary movements in some individuals (although such medications can have side effects and so should not be started lightly), and depression and psychosis can be treated using medication available for such symptoms in the general population. Other drugs may also be used, depending on the individual person’s needs and symptoms. Speech therapy is widely used to improve communication and difficulties with eating and swallowing, and there is a place for input from occupational therapists, psychologists, dentists, social workers and physiotherapists. Indeed, there is accumulating evidence that exercise may be helpful in improving an individual’s function at several stages of the condition.
However, because of the lack of disease-modifying treatments, scientists are making huge efforts to find new treatments. A very important part of this is learning how the genetic error leads to the loss of medium spiny neurons, and the resulting symptoms of the disease, so that new treatments can be properly designed to be most effective. There are a number of clinical trials underway to test out some potential new treatments. However it is important to recognise that it can take a long time for a treatment to go from experimental clinical trials to being available for use in the community. A wide range of treatment strategies are being considered including various drug treatments, gene therapies, exercise and rehabilitation studies and methods to replace lost medium spiny neurons.
Stem cells offer an opportunity to grow and study large numbers of cells in the laboratory. There are several different types of stem cells and scientists are investigating a number of ways they might be used to tackle the challenges of HD:
- Studying and understanding the disease: Researchers can use certain kinds of stem cells to grow large numbers of the types of cells that are damaged in HD. These cells can also be made to carry the genetic fault that causes HD. Then the cells can be used in research to uncover exactly how the disease works and what function(s) the normal gene has in the healthy brain.
- Developing new drugs: the same sorts of cells as above can be used to search for and test new drugs.
- Replacing lost cells: In the future, scientists hope to be able to use stem cells to grow new, healthy MSNs that can be transplanted into patients to replace cells destroyed by the disease.
Understanding the Huntington’s disease and drug discovery
As described above, the Huntingtin gene contains a DNA fragment called a ‘CAG repeat’ – a piece of DNA made up of three units (C, A and G) that appear multiple times in the same order. The healthy gene contains less than 35 CAG repeats, but HD patients have a mutated version that contains too many CAG repeats. These extra copies of the CAG sequence make a faulty, toxic version of the Huntingtin protein. The higher the number of CAG repeats, the more toxic Huntingtin is, and the earlier the symptoms of the disease tend to appear.
Scientists working on the mutant Huntingtin protein are asking the question - how does the change in this one protein cause damage to MSNs? Unfortunately the Huntingtin protein interacts with many other proteins and affects multiple processes in our cells, and so exactly how it contributes to disease progression remains unknown. To study this question, scientists use models of the disease: systems designed to represent HD in the lab so it can be studied outside the patient’s body. Stem cells are being used to develop these models to study the disease and how it progresses.
For example, induced pluripotent (iPS) cells can be made from the muscle or skin of HD patients. They can then be expanded in number many fold in the lab before being treated to persuade them to develop into neurons. One consortium of HD researchers from six American and two European laboratories achieved this and the work was verified independently by a laboratory in California. The iPS cells obtained in this way have the same genetic code as the people affected by HD, and have been made from people with different numbers of CAG repeats. When these different iPS cells are used to make MSNs, the neurons with longer CAG repeats are more vulnerable to different stresses and show many of the characteristics of the disease as it occurs in patients. Researchers hope that these iPS cell systems will now help them uncover exactly how HD works. This approach can in theory provide an almost limitless source of Huntington’s disease MSNs, and much work has, and is still, going on to developing robust systems for making Huntington’s disease iPS cells for such applications.
It is also possible to use these cell models to screen for drugs that may have therapeutic applications. Today’s technology provides mechanical systems that allow researchers to test millions of chemical compounds in a relatively small amount of time to see if they might have a useful effect on cells. These systems are valuable tools for drug discovery, but they require huge numbers of cells of the right type (e.g. neurons) that must have the disease being studied. HD iPS cell models can be used for screening drugs that may be capable of modifying the disease.
Replacing lost cells
Most (though not all) of the symptoms of HD are due to the loss of medium spiny neurons in the brain. Consequently scientists’ efforts have been focused on obtaining new MSNs to replace the damaged ones. In 2000 and 2006 medical doctors reported on transplants of fetal neurons into the brains of a small number of HD patients. The patients whose grafts survived had improvement in both movement and psychological symptoms over a six year period. This provides “proof of concept”, meaning that it demonstrates that transplants of new cells can improve symptoms in HD, but it does not tell us how reliable the procedure is. After the six year period the HD symptoms worsened again, which suggests that the process is not yet optimal and tells us that more work needs to be done using fetal cells to work out how to do this procedure in the best way. It is possible that improved procedures for using these fetal MSNs could help to treat HD and current clinical trials are investigating this idea, but there are some significant scientific and technical problems. For example, the use of tissue taken from aborted fetuses provides only a very limited source of cells which cannot be purified nor improved. Stem cells could provide a valuable alternative.
Researchers hope to use embryonic stem cells or iPS cells as an unlimited source of medium spiny neurons for HD treatment. The iPS cells could be generated from people who don’t have the Huntington’s genetic fault. They could also theoretically be made from the skin of people with Huntington’s where the researchers corrected the genetic problem using gene editing techniques so that the cells produce fully functional Huntingtin protein. MSNs have now been grown from genetically corrected iPS cells and show signs of being healthy, for example in the lab, they are just as resistant to cell death as MSNs from people without HD. Much more research is needed, but this approach could one day mean that a person with Huntington’s could receive a brain cell transplant using “corrected” cells from their own skin.
Both these types of stem cells (embryonic stem cells and iPS cells) have the ability to make all the different cells of the body, so the challenge is to find a way to direct them to make only MSNs. A number of labs have now developed protocols to differentiate neurons that have characteristics of MSNs from human embryonic stem cells. Work is underway to test how closely these neurons resemble genuine MSNs and whether they can improve the symptoms of Huntington’s disease once they have been transplanted into animal models of this disorder. It is hoped that such cells will one day be suitable for transplantation into people with Huntington’s disease.
The use of stem cells for HD research is a relatively recent development. Although stem cells are already beginning to provide valuable tools for studying HD progression and for searching for new drugs, there’s still work to do before treatments involving transplantation of stem cells into patients could be considered. The main questions that must first be answered are:
- How similar are lab-grown MSNs to those found in our brains? Although they have been shown to share some characteristics, they may not be identical and must be better understood so that methods for producing such cells can be improved.
- How long can lab-produced MSNs survive when transplanted into the brain and are they able to do their job there?
- Would these lab-produced MSNs be able to integrate into the damaged adult nervous system, reconnect the lost circuitries and provide a benefit to patients?
- Would transplantation of MSNs be safe for patients, especially in the long term?
So, stem cells cannot be used to treat HD today, but stem cell research is providing useful tools for scientists aiming to develop new approaches for the future. This work is going on in a number of labs internationally and several of them are involved in two European Commission-funded research Consortia: Repair-HD and NeurostemcellRepair.
This factsheet was created by Serafi Cambray and reviewed by Elena Cattaneo.This fact sheet was later reviewed by Anne Rosser.
Lead image of neurons grown from embryonic stem cells by Serafi Cambray. Medium spiny neurons labelled with green fluorescent proteins by Valentina Castiglioni. Human medium spiny neurons grown from embryonic stem cells by Charles Arber. All other images and figures by Serafi Cambray. Figure created using Servier Medical Art.
Last updated: