Access to Regenerative Medicine in the NHS: Regulation and Reimbursement
by Aurélie Mahalatchimy and Alex Faulkner
According to what is defined as a regenerative medicine-based product in law, different regulations, and consequently market access pathways, will apply.
Regenerative medicine based products, which are developed for therapeutic purposes, can be:
- Medical Devices (Regulations (EU) 2017/745 and 2017/746 which recently replace Directives 90/385/EEC, 93/42/EEC, and 98/79/EC)
- Medicinal products (Mainly Directive 2001/83/EC). In that case, they will generally be Advanced Therapy Medicinal Products (ATMPs), i.e. gene therapy, cell therapy, tissue engineered medicinal products or combination of these products with medical devices (Regulation (EC) No 1394/2007)
- Tissues and cells (Mainly Directives 2004/23/EC, 2006/86/EC, 2006/17/EC)
- Blood and blood components (Mainly Directive 2002/98/EC)
The law provides legal definitions for each type of these products. Moreover, different criteria have been established to distinguish which type of regenerative medicine based products are being developed and consequently which set of regulation applies both for market access and for reimbursement decisions. The main criteria are:
- The scale of development of the product
- Is the product developed at the ‘industrial’ level or not?
- The degrees of manipulation of tissues and cells
- Have tissues and cells been substantially manipulated or not?
- Are tissues and cells used for the same essential functions in the recipient as in the donor?
Despite these tools (legal definitions and criteria) that have been developed, it remains very difficult to know exactly what kind of regenerative medicine based product is being developed according to the law. That is why producers are recommended to contact regulators (either or both at the national or European levels) and reimbursement bodies as early as possible. In such context, there exists a specific committee at the European Union level and specific offices in the UK to help on this issue:
- The Committee for Advanced Therapies at the European Medicines Agency regarding the marketing authorisation of Advanced Therapy Medicinal Products in the European Union
- The “One- stop- shop” at the Medicines and Healthcare products Regulatory Agency (MHRA) in the UK for the commercialisation of other products mainly in the UK
- The Office for Market Access of the National Institute for Health and Care Excellence (NICE) for the decisions on reimbursement.
Advanced Therapy Medicinal Product is the legal category the most supported by European Union law both for economics and public health objectives. However, being an Advanced Therapy Medicinal Product (ATMP) does not mean there is one regulatory pathway only for commercialisation. Indeed, three regulatory pathways can be used.
- If the ATMP is developed at the industrial scale, it will be entirely covered by European Union law (Mainly Directive 2001/83/EC and Regulation (EC) No 1394/2007). Notably, it implies that the ATMP shall be manufactured in a licenced establishment in accordance with Good Manufacturing Practices and that Good Clinical Practices shall be respected for clinical trials. It also needs a centralised European marketing authorisation to be commercialised in the European Union (including in the UK). The European Commission grants the centralised European marketing authorisation after scientific assessment and opinion of the European Medicines Agency. For ATMP, the Committee for Advanced Therapies has to be involved (it gives an opinion) within the European Medicines Agency.
- If the ATMP is not developed at the industrial scale, national laws will cover it. In the UK, two regulatory pathways exist: the Hospital Exemption pathway (that also exists in other European Member States) and the Specials pathway (that is specific to the UK) (See table 1 below). In both case, the MHRA will be the competent authority.
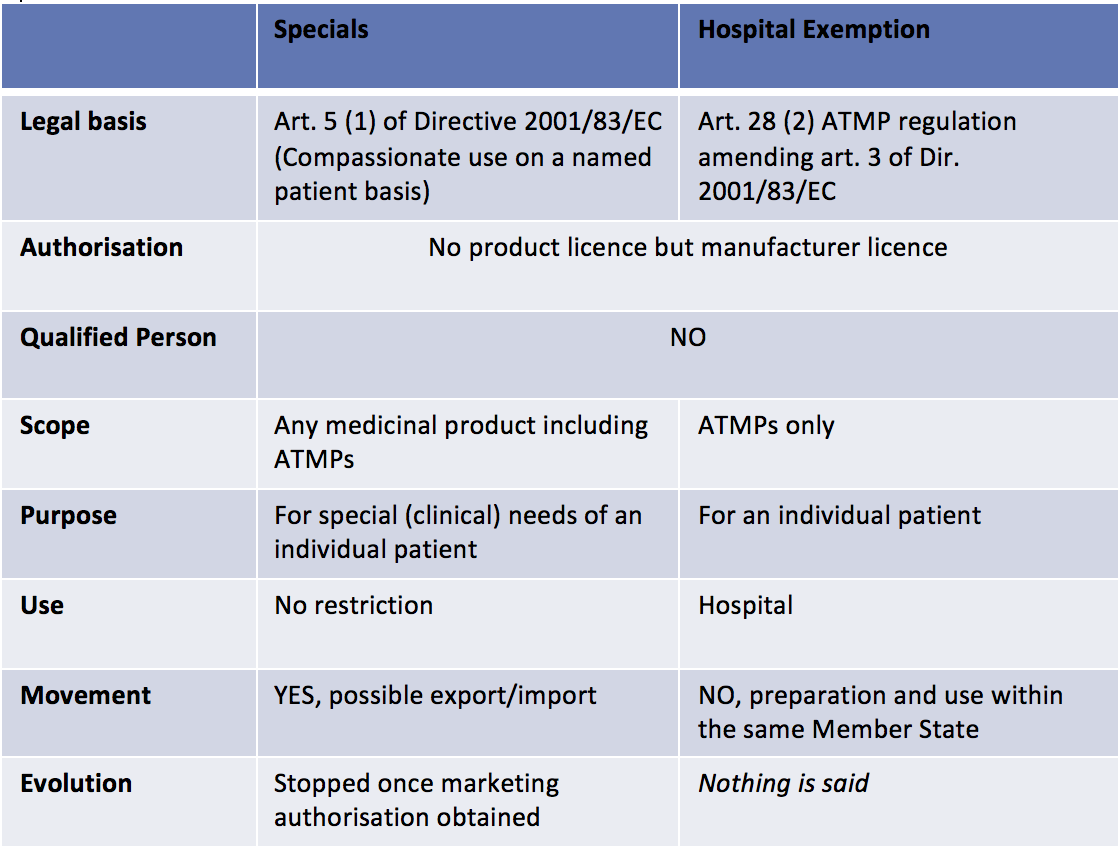
When Regenerative Medicine based products are ATMPs developed at the industrial scale, many incentives apply: Committee for Advanced Therapies’ recommendations on the classification as ATMPs, specific procedure to certify quality and non-clinical information to facilitate the development of ATMPs through partnerships between the academic world, SMEs and big pharmaceutical companies, a 90% reduction for SMEs and a 65% reduction for other applicants with regard to the fees payable to European Medicines Agency for the provision of scientific advice.
Moreover, ATMPs developed at the industrial scale can also be recognised as orphan drugs (the “orphan medicinal product” designation to be obtained from the committee for orphan medicinal products at the European Medicines Agency). In that case, they can also benefit from incentives provided for orphan drugs: ten-year market exclusivity period, assistance with the elaboration of protocols with a partial or total waiver of the fees payable to European Medicines Agency, eligibility for European Union and Member States incentives to support R&D for orphan drugs, total or partial reduction of the fees due to European Medicines Agency. Indeed, among the 8 ATMPs that have been authorised since the adoption of Regulation (EC) No 1394/2007 on ATMPs, half of them are orphan drugs.
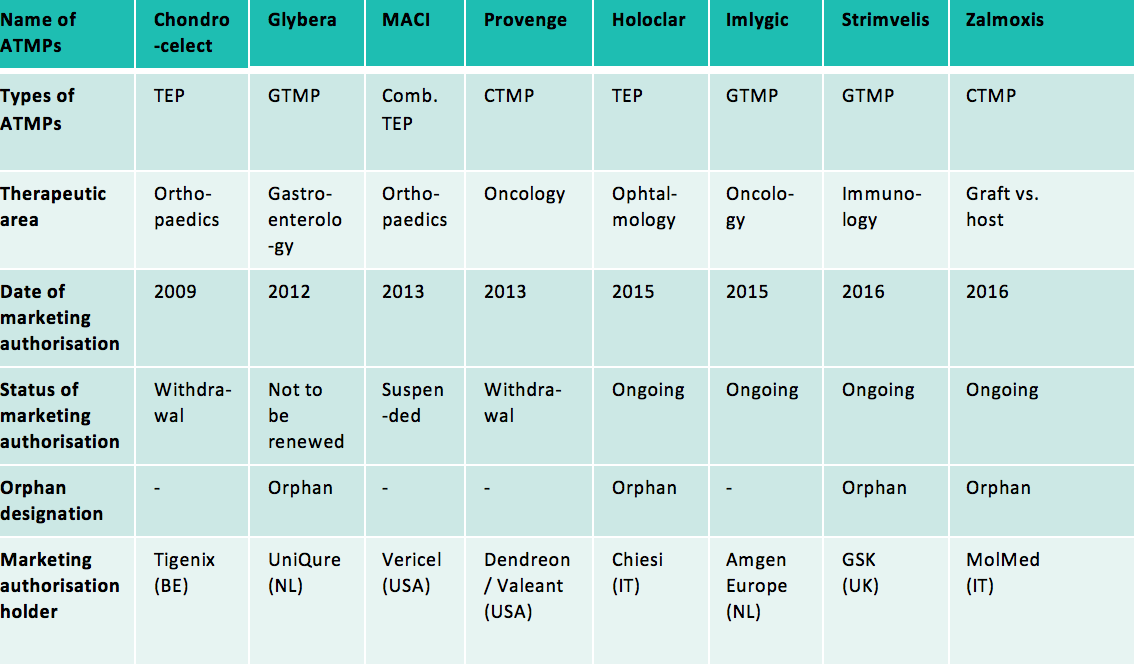
Abbreviations used: GTMP: Gene Therapy Medicinal Product; CTMP: Cell Therapy Medicinal Product; TEP: Tissue Engineered Product; Comb.: Combined
Among these 8 authorised ATMPs, half of them are no longer on the market (or will not be on the market by the end of 2017) at the request of their marketing authorisation holders (Chondrocelect, Glybera, Provenge) or because of lack of authorised manufacturing facility in the European Union (MACI).
Thus, ATMPs are experiencing market failures. One of the reasons is that they are high cost and they are not necessarily reimbursed by national health insurance in the Member States of the European Union. Indeed, ATMPs as other Regenerative Medicine based products raise several reimbursement issues especially: low evidence of long term cost/effectiveness data, high cost, limited health resources, raised concerns about lack of flexibility of reimbursement regulatory pathways.
In the UK, there has been much thinking around regenerative medicine reimbursement issues from various actors, notably in public reports such as:
- Regenerative Medicine Expert Group. Building on our own potential: a UK pathway for regenerative medicine (2015). (Accessed July 2016)
- NICE. Exploring the assessment and appraisal of regenerative medicines and cell therapy products (2016).(Accessed July 2016)
- Advanced Therapies Manufacturing Task Force Action Plan. Retaining and attracting advanced therapies manufacture in the UK (2017).
Various stakeholders debate the different values of regenerative medicine.
In such context, new flexibilities have been introduced in addition to those previously mentioned (MHRA ‘One Stop shop’ and NICE Office for Market Access) focusing on collaborations between institutions:
- New regulatory tools have been established at the European level either to fill in the gap between entering the market and being reimbursed by national health insurances (Adaptive Pathways) or to support medicines development, including ATMPs (Enhanced early dialogue to facilitate accelerated assessment for PRIority Medicines (PRIME)). The criteria for such schemes are unmet medical needs; schemes include provision for enhanced interaction between developers and regulators and earlier involvement of health technology assessment agencies.
- In the UK, the MHRA has introduced the ‘Promising Innovative Medicine’ (PIM) designation as an incentive, under an Early Access to Medicines Scheme (EAMS), where producers have reduced data requirements and provide the therapy to small number of patients at no cost. Moreover, the UK Government commissioned the Accelerated Access Review (AAR)’s report that makes recommendations for a proposed approach for patients’ quicker access to innovative medicines, medical devices, digital products and emerging forms of treatment.