Chorea Huntington: Hilfe durch Stammzellen?
Die Chorea Huntington (Huntington-Krankheit) ist eine schwere erbliche neurodegenerative Erkrankung, an der in den USA, Europa und Australien etwa einer von 10.000 Menschen (ca. 8.000 Patienten in Deutschland) leidet. Typischerweise verursacht sie unfreiwillige Gesichts- und Körperbewegungen sowie Demenz. Die Symptome verschlimmern sich mit der Zeit, bis die erkrankte Person völlig auf die Hilfe anderer angewiesen ist. Bis heute gibt es keine wirksame Therapie. Wie könnten Stammzellen neue Behandlungsformen erschließen?
Was wissen wir? ▼
Die Chorea Huntington (Huntington-Krankheit) ist eine Erbkrankheit, bei der die striatalen Projektionsneuronen (im Englischen bezeichnet als „Medium Spiny Neurons“ bzw. MSN) im Gehirn zerstört werden, was zu emotionalen Veränderungen, geistigem Verfall und Bewegungsstörungen führt.
Die Symptome treten häufig im Alter von 35 bis 50 Jahren auf, können aber auch schon früher beginnen.
Chorea Huntington wird von einer CAG-Sequenz der DNA verursacht, die im Huntington-Gen über 40 Mal wiederholt wird. Bei Menschen mit mehr als 35 Wiederholungen besteht ein erhöhtes Erkrankungsrisiko.
Es gibt derzeit keine Therapien der Chorea Huntington.
Stammzellen haben als Mittel zur Erforschung vieler Aspekte der Chorea Huntington einen unschätzbaren Wert. Dies reicht von der Untersuchung, warum bei Chorea Huntington MSN zugrunde gehen, bis zur Testung neuer Behandlungsformen.
The German article translates "medium spiny neurons" in a very strange way which I cannot verify in the internet. Most of the references use the English abbreviation with an explanation of what it means in English and then use terms such as the chosen one "striatale Projektionsneuronen".
Was untersuchen Forscher? ▼
Das Huntington-Gen bildet ein Protein, das wichtig für Neuronen ist. Doch die Wissenschaftler versuchen immer noch zu verstehen, was dieses Protein in Zellen tut und warum zu viele CAG-Wiederholungen dazu führen, dass MSN absterben.
Die Forscher verwenden aus induzierten pluripotenten Stammzellen (iPS) gezüchtete MSN als Modell der Chorea Huntington und beobachten daran das Fortschreiten der Erkrankung. iPS werden auch zum raschen Testen von neuen Arzneimitteln, Gentherapien und anderen Behandlungen von Chorea Huntington verwendet. Das geht schneller als zu der Zeit, in der iPS noch nicht verfügbar waren.
In Studien wird untersucht, wie neurale Stammzellen (eine im Gehirn vorkommende Stammzellart) ins Gehirn von Patienten transplantiert werden könnten, um das Gehirn zu reparieren und möglicherweise MSN und andere benötigte Gehirnzellen nachzuzüchten.
Was sind die Herausforderungen? ▼
Gegenwärtig gibt es keine zugelassenen Stammzelltherapien für Chorea Huntington. Die Transplantation neuraler Stammzellen wurde in mehreren wissenschaftlichen Studien zur Behandlung von Patienten mit Chorea Huntington untersucht, doch diese Therapien müssen noch strenge klinische Studien durchlaufen, um ihre Sicherheit und Wirksamkeit unter Beweis zu stellen.
Ethische und zuverlässige Quellen für neurale Stammzellen müssen erschlossen werden. iPS und embryonale Stammzellen sind unter Umständen in der Lage, große Mengen an neuralen Stammzellen hervorzubringen. Doch diese Zellen müssen identisch, berechenbar und sicher sein. Ohne ausreichende Tests könnten pluripotente Stammzellen Krebs oder andere lebensbedrohliche Komplikationen hervorrufen.
- Was ist die Chorea Huntington?
- HD und „CAG-Wiederholungen“
- Wie wird die Chorea Huntington heute behandelt?
- Wie könnten Stammzellen helfen?
- Aktuelle Forschung
- Sind Stammzellen schon jetzt zur Behandlung von HD einsetzbar?
- Erfahren Sie mehr
- Find out more (English)
- Danksagungen und Quellenangaben
Die Chorea Huntington (abgekürzt HD für Huntington's Disease) greift hauptsächlich Nervenzellen im Gehirn an, die so genannten mittleren stacheligen Neurone (MSN, engl. Medium Spiny Neurons). MSNs nehmen Informationen von anderen Nervenzellen im Gehirn entgegen und koordinieren sie, um die Bewegungen von Körper, Gesicht und Händen zu steuern.
Bei der HD sind viele MSN beschädigt oder zerstört. Anscheinend sind auch andere Arten von Nervenzellen im Gehirn wie kortikale Nervenzellen betroffen. Die ersten Symptome treten in der Regel im Alter von 35-50 Jahren auf, meist in Form schwach spastischer Muskelzuckungen in Gesicht und Gliedern. Mit dem Fortschreiten der Krankheit werden diese Spasmen deutlicher und häufiger, und weitere Symptome kommen hinzu. Es ist je nach Patient unterschiedlich, aber mögliche Symptome sind Schwierigkeiten beim Sprechen und Schlucken, Demenz oder Konzentrationsstörungen.
HD ist eine Erbkrankheit. Kinder erkrankter Eltern erben mit 50%iger Wahrscheinlichkeit den Gendefekt, der die Krankheit auslöst. Dieser Defekt betrifft ein Gen, auf dem die Information für das Protein Huntingtin liegt. Durch dieses defekte Gen produziert der Körper eine fehlerhafte, toxische Version des Huntingtin-Proteins. Das führt letztendlich zum Verlust von MSNs und anderen Nervenzellen.
Das DNA-Alphabet besteht aus vier Buchstaben: C, A, G und T. Dies sind die vier organischen Basen der Nukleinsäure, welche in unterschiedlicher Reihenfolge angeordnet die DNA-Stränge bilden. In seiner nicht krankhaften Version enthält das Huntingtin-Gen bis zu 35 Wiederholungen der Sequenz CAG. In dieser Form wirkt sich das Gen positiv auf die Nervenzellen aus. Wissenschaftler entdeckten, dass das Gen zuerst ohne jegliche CAG-Wiederholungen auftrat, und zwar bei einer Amöbe, der Dictyostelium discoideum. Mit der Entwicklung neuer Arten entstanden die CAGs, und je komplexer das Nervensystem einer Art, desto mehr davon traten auf. Es gibt Anzeichen, dass die Anzahl der CAG-Wiederholungen im normalen Gen mit der Menge an Nervenzellen korreliert sein könnte. Bei gesunden Menschen hieße dies: Je mehr CAG auf einem Gen (bis maximal 35), desto mehr graue Substanz im Gehirn. Die CAG-Wiederholungen sind also möglicherweise korreliert mit der Komplexität und Funktion des Gehirns, sowohl über die Evolution hinweg als auch beim Menschen. Dies ist allerdings noch unbewiesen und derzeit nur eine interessante Hypothese. Sicher ist jedoch, dass mehr als 40 CAG-Wiederholungen beim Menschen die Huntington-Krankheit auslösen.
Es gibt bisher keine wirksame Therapie, mit der sich der Krankheitsverlauf bei HD aufhalten oder umkehren ließe. Man konzentriert sich stattdessen auf die Linderung der Symptome. Zur Behandlung der unwillkürlichen Bewegungen werden meist Tetrabenazin, Benzodiazepine und Antipsychotika eingesetzt. Letztere können auch bei einigen der möglichen psychischen Symptome helfen. Logopädie wird häufig genutzt, um die kommunikativen Fähigkeiten zu verbessern und Ess- und Schluckstörungen entgegenzuwirken. Auch andere Medikamente können zum Einsatz kommen, je nach individuellen Bedürfnissen und Symptomen.
Diese starken Medikamente haben leider oft Nebenwirkungen und können weder die Schäden reparieren, noch den Krankheitsverlauf stoppen. Daher arbeiten Forscher mit voller Kraft an neuen Verfahren, um die Krankheit an der Wurzel zu packen und verlorene mittlere stachelige Neurone zu ersetzen.
Zwar ist bekannt, dass HD durch einen Defekt des Gens ausgelöst wird, welches das Protein Huntingtin bildet. Unklar jedoch ist noch, wie der Gendefekt zum Verlust der MSNs und damit zu den Krankheitssymptomen führt. Wäre dieser Prozess bekannt, ließen sich vielleicht neue Behandlungsoptionen ableiten. Die Wissenschaftler hoffen auch, Wege zum Ersatz verloren gegangener MSN bei Patienten zu finden.
Mithilfe von Stammzellen lassen sich große Mengen von Zellen im Labor kultivieren und untersuchen. Es gibt verschiedene Arten von Stammzellen. In der Forschung untersucht man derzeit mehrere Wege, diese irgendwann zur Bewältigung der Herausforderungen bei HD zu nutzen:
- Erforschung und Verständnis der Krankheit: Mit bestimmten Arten von Stammzellen lassen sich große Mengen derjenigen Zellen kultivieren, die bei HD beschädigt werden. Diese können dann in der Forschung verwendet werden, um festzustellen, wie genau die Krankheit abläuft und welche Funktion(en) das normale Gen im menschlichen Gehirn erfüllt.
- Ersatz verlorener Zellen: Für die Zukunft hoffen Wissenschaftler, mithilfe von Stammzellen neue, gesunde MSNs zu erzeugen. Diese würden dann den Patienten transplantiert, um durch die Krankheit zerstörte Zellen zu ersetzen.
- Entwicklung neuer Medikamente: Bestimmte Arten von Stammzellen könnten dazu dienen, MSNs mit dem Gendefekt zu züchten, der die Chorea Huntington auslöst. Diese Zellen wären dann sehr wertvoll bei der Suche nach neuen medikamentösen Wirkstoffen und ihrer Erprobung.
Ersatz verlorener Zellen
Die meisten (aber nicht alle) Symptome bei HD sind auf den Verlust der mittleren stacheligen Neurone (MSN) im Gehirn zurückzuführen. Deshalb konzentrierte sich die Forschung bislang darauf, neue MSNs zu gewinnen, um die zerstörten zu ersetzen. Im Jahr 2000 verpflanzten Ärzte fetale Nervenzellen in das Gehirn einiger weniger HD-Patienten. Dies führte zu einer kurzfristigen Besserung der Bewegungsstörungen und der psychischen Symptome, aber zwei Jahre nach dem Eingriff verschlechterten sich die Symptome wieder. Es ist denkbar, dass verbesserte Verfahren zur Anwendung dieser fetalen MSNs bei der Behandlung von HD helfen können, und dies wird in aktuellen klinischen Studien untersucht, aber es gibt einige große wissenschaftliche und technische Probleme: zum Beispiel lässt sich aus dem Gewebe abgetriebener Föten nur eine sehr begrenzte Menge an Zellen gewinnen, die sich auch weder reinigen noch aufbereiten lässt. Stammzellen könnten hier eine gangbare Alternative darstellen.
Wissenschaftler hoffen auf embryonale Stammzellen oder induzierte pluripotente Stammzellen als unbegrenzte Quelle für mittlere stachelige Neurone zur Behandlung von HD. Diese beiden Arten von Stammzellen können alle im Körper vorkommenden Zelltypen hervorbringen. Die Herausforderung besteht also darin, sie dazu zu bekommen, nur MSNs zu erzeugen. Die ersten im Labor kultivierten Nervenzellen, die MSNs ähnelten, wurden 2008 aus embryonalen Stammzellen gewonnen. Bei der Verpflanzung in Nager jedoch ließen sie Tumore wachsen. 2012 dann erzeugten zwei Labore MSNs aus menschlichen embryonalen Stammzellen auf eine Weise, die keine Tumorbildung auslöste. Verpflanzt man diese im Labor gezüchteten MSNs in Nager, die an einer Form von HD leiden, zeigt sich eine gewisse Besserung. In einem Fall konnte auch gezeigt werden, dass die Nervenzellen eine Reihe von Schlüsseleigenschaften besaßen, wie sie auch echte MSNs im menschlichen Gehirn haben. Es ist noch viel Forschung nötig, um Qualität und Quantität der gewonnenen Nervenzellen zu steigern und festzustellen, ob dieser Ansatz sich eines Tages gefahrlos und wirksam auf menschliche Patienten übertragen lässt.
Einige jüngere Studien haben ergeben, dass es auch möglich ist, induzierte pluripotente Stammzellen (iPS Zellen) von an Chorea Huntington erkrankten Patienten zu gewinnen. Ein Zusammenschluss von HD-Forschern aus sechs amerikanischen und zwei europäischen Labors entnahm HD-Patienten Hautzellen und reprogrammierte sie so, dass sie iPS Zellen erzeugten. Auch ein Labor in Kalifornien kam eigenständig so weit. Die erhaltenen iPS Zellen haben denselben genetischen Code wie der Patient, also auch den HD auslösenden genetischen Defekt, und zeigen einige der typischen Krankheitsmerkmale. Die Forscher korrigierten den genetischen Defekt im Labor, sodass die Zellen wieder korrekt funktionierendes Huntingtin-Protein produzierten. Die aus diesen genetisch reparierten iPS Zellen gezüchteten MSNs zeigten Anzeichen von Gesundheit: Im Labor sind sie ebenso widerstandsfähig gegen Zelltod wie MSNs von Menschen ohne HD. Es ist noch viel Forschung nötig, aber dieser Ansatz könnte eines Tages ein wichtiges Werkzeug werden, um Zellersatztherapien bei HD durchzuführen.
Screening und Entdeckung von Wirkstoffen
Die moderne Technologie hält mechanische Anlagen bereit, mit denen Forscher in relativ kurzer Zeit Millionen chemischer Zusammensetzungen testen können, um zu sehen, ob sie vielleicht nützliche Auswirkungen auf Zellen haben. Diese Anlagen sind sehr wertvoll bei der Suche nach neuen Medikamenten, aber es gibt einen Haken: Riesige Mengen von Zellen der richtigen Art (z.B. Nervenzellen) sind erforderlich, und diese Zellen müssen die zu untersuchende Krankheit aufweisen. Es ist unmöglich, von Huntington befallene MSNs direkt von Patienten zu gewinnen, denn sie müssten chirurgisch aus dem Gehirn entnommen werden. Mit iPS Zellen aus Muskel- oder Hautgewebe von HD-Patienten dagegen ließe sich vielleicht eine unbegrenzte Zahl von Huntington-MSNs gewinnen und so das Problem lösen. Ein Großteil der Forschung konzentriert sich derzeit auf die Entwicklung verlässlicher Systeme zur Erzeugung von an Huntington erkrankten iPS Zellen, um damit nach neuen Wirkstoffen zu forschen.
Erforschung des Krankheitsverhaltens
Mutationen (oder Veränderungen) des Gens, das für Huntingtin-Protein codiert, wurden erstmals 1993 mit der Chorea Huntington in Verbindung gebracht. Das Huntingtin-Gen enthält einen DNA Abschnitt, welcher aus drei Einheiten (C, A und G) besteht, die mehrfach in derselben Reihenfolge erscheinen („CAG-Wiederholungen“). Das gesunde Gen enthält weniger als 35 CAG-Wiederholungen. Die mutierte Version der HD-Patienten weist mehr als 35 CAG-Wiederholungen auf, in sehr seltenen Fällen sogar bis zu 250. Der Bereich von 36 – 40 CAG-Wiederholungen birgt ein erhöhtes Risiko für die Erkrankung. Bei mehr als 40 CAG-Wiederholungen bricht Chorea Huntington sicher aus. Diese überzähligen Kopien der Sequenz CAG produzieren eine fehlerhafte, toxische Version von Huntingtin. Je mehr CAG-Wiederholungen, desto toxischer ist das Huntingtin, und desto früher treten die ersten Krankheitsanzeichen auf.
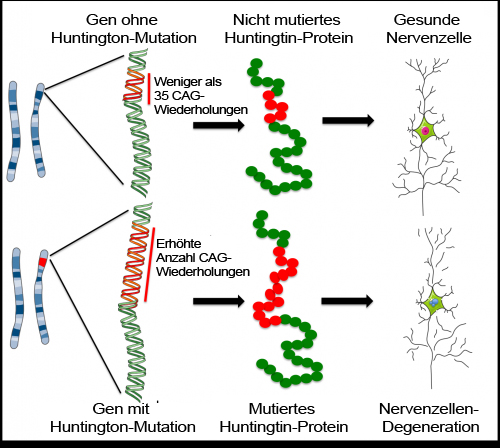
Forscher, die sich mit dem mutierten Huntingtin-Protein beschäftigen, konzentrieren sich auf die Aufklärung der Mechanismen, mit denen es zur Krankheit beiträgt: Wie führt eine Veränderung an diesem Protein zu Schäden an anderen Proteinen und den MSNs? Das Huntingtin Protein interagiert mit vielen anderen Proteinen und beeinflusst zahlreiche Prozesse in den Zellen. Wie genau es die Krankheitsentwicklung beeinflusst, ist noch unbekannt. Um das zu untersuchen, verwendet man Krankheitsmodelle: Systeme, die HD im Labor darstellen können, um sie außerhalb der Körper von Patienten zu studieren. Die heutigen Modelle verwenden Nagetiere oder genetisch veränderte Zellen, aber es gibt noch kein hinreichendes Modell, welches die Krankheit exakt abbilden könnte. Keines der Modelle reproduziert die Verknüpfung zwischen der Anzahl der CAG-Wiederholungen und der Toxizität des Proteins, wie man sie bei Patienten findet.
Stammzellen könnten dazu dienen, ein genaueres Modell zur Erforschung der Mechanismen und des Verlaufs der HD zu schaffen. Es wurden bereits iPS Zellen von Patienten mit unterschiedlich vielen CAG-Wiederholungen kultiviert. Werden aus diesen Zellen MSNs erzeugt, so sind diese Nervenzellen mit mehr CAG-Wiederholungen anfälliger für verschiedene Stressreize und zeigen mehr Anzeichen der Krankheit, wie sie auch bei den Patienten auftreten. Forscher hoffen, mithilfe dieser iPS Zellsysteme herauszufinden, wie HD genau funktioniert.
Die Nutzung von Stammzellen in der Forschung zu HD ist eine sehr junge Entwicklung. Sie erweisen sich zwar bereits jetzt schon als wertvolle Werkzeuge zur Erforschung des Krankheitsverlaufs und der Suche nach neuen Wirkstoffen, aber es ist noch ein langer Weg, bis Behandlungen mit Zellverpflanzungen in Patienten als zuverlässiger Ansatz bezeichnet werden können. Zuerst sind folgende Hauptfragen zu beantworten:
- Wie ähnlich sind die im Labor kultivierten MSNs denjenigen im menschlichen Gehirn? Es wurde zwar gezeigt, dass einige Merkmale übereinstimmen, jedoch sind sie höchstwahrscheinlich nicht identisch. Hier muss noch mehr geforscht werden, um Verfahren zur Beurteilung und Aufrechterhaltung der Qualität von Laborzellen entwickeln zu können.
- Wie lange können im Labor gezüchtete MSNs überleben, wenn sie ins Gehirn transplantiert werden, und können sie dort ihre Aufgaben erfüllen?
- Sind Laborzellen in der Lage, sich in das geschädigte ausgewachsene Nervensystem zu integrieren, gestörte Schaltkreise neu zu verbinden und den Patienten zu helfen?
- Wäre eine Transplantation von MSNs für Patienten gefahrlos, vor allem über längere Zeit?
Stammzellen sind demnach heute noch nicht zur Behandlung von HD einsetzbar. Die Stammzellforschung liefert jedoch wichtige Werkzeuge, um neue Methoden für die Zukunft zu entwickeln.
EuroStemCell Factsheet zu embryonalen Stammzellen
EuroStemCell Factsheet zu iPS Zellen und Reprogrammierung
European Huntington's Disease Network (mit deutschen Inhalten)
Deutsche Huntington Hilfe e.V.
Schweizerische Huntington Vereinigung (SHV)
Österreichische Huntington-Hilfe
Fetale Stammzellen: Hoffnung für Hirnerkrankungen? – wissenschau.de
This factsheet was created by Serafi Cambray and reviewed by Elena Cattaneo.This fact sheet was later reviewed by Anne Rosser.
Dieser Artikel wurde von Serafi Cambray verfasst und von Elena Cattaneo rezensiert.
Die deutsche Übersetzung wurde im Auftrag des German Stem Cell Network (GSCN) erstellt und von Erich Wanker rezensiert.
Hauptbild von Neuronen, die aus embryonalen Stammzellen hergestellt wurden, von Serafi Cambray. Mittlere stachelige Neurone, mit grün fluoreszierendem Protein markiert von Valentina Castiglioni. Menschliche mittlere stachelige Neurone, die aus embryonalen Stammzellen hergestellt wurden, von Charles Arber. Alle anderen Bilder und Abbildungen von Serafi Cambray. Die Abbildung “HD und die Gene” wurde mit Servier Medical Art hergestellt.
Zuletzt aktualisiert: