Choroba Huntingtona: Jak mogą pomóc komórki macierzyste?
Choroba Huntingtona jest wyniszczającą, dziedziczną chorobą neurodegeneracyjną dotykającą około 5 na 100.000 ludzi w USA, Europie i Australii. Najczęściej powoduje niekontrolowane ruchy twarzy i ciała oraz demencję. Z czasem symptomy choroby pogarszają się, aż dotknięta chorobą osoba pozostaje całkowicie zależna od pomocy innych. Obecnie nie są dostępne żadne skuteczne terapie. W jaki sposób badania nad komórkami macierzystymi mogłyby prowadzić do odkrycia nowych metod leczenia?
Co wiadomo? ▼
Choroba Huntingtona (ang.: HD) to genetyczna choroba dziedziczna, która niszczy średnie neurony kolczaste (ang.: MSNs) w mózgu powodując zmiany emocjonalne, zakłócenia procesów intelektualnych oraz zaburzenia ruchowe.
Symptomy często pojawiają się u osób pomiędzy 35-50 rokiem życia.
HD występuje na skutek zbyt dużej (>40) ilości powtórzeń sekwencji ‘CAG’ w genie huntingtiny.
Obecnie nie ma skutecznej terapii leczenia choroby Huntingtona.
Komórki macierzyste są nieocenionym narzędziem w badaniu wielu aspektów HD, od zrozumienia jak dochodzi do śmierć komórek MSN po testowanie nowych terapii.
Co analizują badacze? ▼
Gen huntingtiny odpowiada za powstanie białka ważnego dla neuronów. Naukowcy ciągle starają się zrozumieć, za co odpowiada to białko i dlaczego zbyt duża ilość powtórzeń ‘CAG’ powoduje śmierć komórek MSN.
Używając MSN stworzonych z indukowanych pluripotencjalnych komórek macierzystych (ang.: iPSC) można obserwować jak postępuje choroba HD. iPSC są także używane do testowania nowych leków lub terapii szybciej niż było to możliwe przed pojawieniem się iPSC.
Nowe badania dotyczą możliwości przeszczepiania nerwowych komórek macierzystych do mózgu osoby chorej, aby odnowiły zniszczone komórki MSN.
Na czym polegają trudności? ▼
Obecnie żadna metoda leczenia HD komórkami macierzystymi nie jest zatwierdzona. Przeszczepy nerwowych komórek macierzystych testowane są w leczeniu osób z HD, ale musza przejść jeszcze wiele badań, aby wykazać swoją skuteczność i bezpieczeństwo.
Etyczne i pewne źródła nerwowych komórek macierzystych muszą zostać opracowane. iPSC i embrionalne komórki macierzyste mogą wytwarzyć większa ilość nerwowych komórek macierzystych, ale komórki te muszą być bezpieczne, przewidywalne. Jeśli nie będą odpowiednio przetestowane PSC mogą przekształcić się w nowotwory lub powodować inne groźne komplikacje.
- Czym jest choroba Huntingtona?
- Choroba Huntingtona i ‘powtórzenia CAG’
- Jak obecnie leczona jest choroba Huntingtona?
- Jak mogą pomóc komórki macierzyste?
- Obecne badania
- Czy komórki macierzyste mogą być obecnie używane do leczenia HD?
- Dowiedz się więcej
- Find out more (English)
- Źródła i odwołania
Choroba Huntingtona (ang. Huntington’s Disease; HD) głównie dotyka komórek nerwowych w mózgu zwanych średnimi neuronami kolczastymi (ang. medium spiny neurons; MSNs). Komórki te otrzymują i zarządzają informacjami od innych neuronów w mózgu dotyczącymi kontrolowania poruszania się, procesów intelektualnych oraz emocji. Chociaż najbardziej charakterystyczne zaburzenia ruchowe w chorobie Huntingtona to mimowolne, gwałtowne ruchy zwane pląsawicą, choroba ta powoduje również szereg innych zaburzeń ruchowych włącznie ze spowolnieniem ruchów, dystonią (ruchy bardziej wydłużone i powodujące wykrzywienia w porównaniu z pląsawicą), trudnościami z koordynowaniem precyzyjnymi ruchami oraz zaburzeniami równowagi. Zaburzenia ruchowe mogą dotykać różnych części ciała takich jak kończyny, twarz, oczy oraz mięśnie kontrolujące mowę i przełykanie. Zaburzenia ruchowe są najbardziej charakterystycznymi objawami choroby, ale zakłócenia procesów intelektualnych i emocji często odpowiadają za kalectwo.
W chorobie HD duża ilość neuronów MSN jest uszkadzana lub niszczona. Inne typy neuronów w mózgu też mogą być dotknięte chorobą np. neurony kory mózgowej (ang. cortical neurons). Pierwsze symptomy zaburzeń ruchowych u ludzi występują najczęściej pomiędzy 35- a 50-tym rokiem życia, chociaż symptomy te mogą pojawić się znacznie wcześniej lub później, a pewne niewielkie problemy związane z emocjami czy myśleniem mogą wystąpić na długo przed zaburzeniami ruchowymi. Ludzie mogą być dotknięci chorobą w różny sposób, w szczególności we wczesnych latach choroby, ale u wszystkich osób objawy choroby będą postępować w ciągu kolejnych dwóch lub trzech dekad.
HD jest chorobą dziedziczną. Dzieci, których rodzice dotknięci są chorobą, mają 50% szans na odziedziczenie wady genetycznej, która powoduje chorobę. Ta wada pojawia się w genie, który koduje białko zwane białkiem huntingtiny. Wadliwy gen powoduje, że organizm produkuje toksyczną wersję białka huntingtiny, co ostatecznie prowadzi do utraty neuronów MSN, a także innych typów neuronów.
„Alfabet DNA” składa się z czterech liter: C, A, G i T. Są to cztery jednostki kwasu nukleinowego, ułożone w różnej kolejności, budujące nić DNA. Prawidłowa wersja genu huntingtiny zawiera do 35 powtórzeń sekwencji CAG. Więcej niż 40 powtórzeń powoduję, że u danej osoby w jakimś momencie życia rozwinie się choroba Huntingtona (chociaż wiek w którym choroba może się rozwinąć jest trudny do przewidzenia). 36-39 powtórzeń stanowi szarą strefę; u osoby z tą ilością powtórzeń może rozwinąć się choroba Huntingtona w trakcie ich życia, ale nie musi (ryzyko wzrasta wraz ze wzrostem ilości powtórzeń).
Co ciekawe, naukowcy odkryli, że po raz pierwszy gen huntingtiny pojawił się u ameby zwanej Dictyostelium descoideum i nie posiadał w ogóle powtórzeń. Wraz z ewolucją nowych gatunków pojawiały się powtórzenia CAG w genie huntingtiny, a ich liczba rosła u gatunków z coraz bardziej skomplikowanym układem nerwowym sugerując, że dłuższe powtórzenia CAG dają neuronom dodatkową korzyść.
Obecnie nie ma żadnej skutecznej metody, aby zatrzymać lub odwrócić postęp choroby, ale istnieją sposoby, by pomóc złagodzić objawy choroby. Na przykład niektóre dostępne leki mogą zahamować mimowolne ruchy u niektórych chorych (chociaż takie leczenie może mieć skutki uboczne i nie powinno być pochopnie stosowane), a depresja i psychoza mogą być leczone ogólnie dostępnymi środkami na podobne dolegliwości. Inne leki też mogą być używane w zależności od potrzeb i symptomów danej osoby. Terapia mowy jest powszechnie stosowana, aby poprawić komunikację i problemy z jedzeniem oraz przełykaniem, pomóc mogą również asystenci rodzinni, psycholodzy, dentyści, pracownicy socjalni i fizjoterapeuci. Rzeczywiście, istnieją dowody na to, że ćwiczenia mogą być pomocne w poprawie indywidualnych funkcji na różnych etapach choroby.
W związku z tym, że brakuje metod leczenia choroby, naukowcy wkładają wiele wysiłku w odkrycie nowych terapii. Ważną częścią tych poszukiwań jest odkrycie w jaki sposób błąd genetyczny prowadzi do utraty średnich neuronów kolczastych, i w rezultacie do symptomów choroby, co pozwoli na odpowiednie zaprojektowanie bardziej skutecznych metod leczenia. Obecnie prowadzonych jest wiele prób klinicznych w celu przetestowania potencjalnie nowych terapii. Jednak należy pamiętać, że może minąć dużo czasu od eksperymentalnej próby klinicznej do gotowej terapii ogólnie dostępnej dla społeczeństwa. Rozważanych jest wiele różnych strategii leczenia włącznie z leczeniem różnymi typami leków, terapią genową, ćwiczeniami i rehabilitacją oraz metodami zastępowania utraconych średnich neuronów kolczastych.
Komórki macierzyste dają możliwość hodowania i badania dużej liczby komórek w laboratorium. Istnieje wiele różnych typów komórek macierzystych, a naukowcy sprawdzają wiele różnych możliwości, aby pokonać trudności związane z HD:
- Badanie i zrozumienie choroby: Naukowcy mogą użyć danego typu komórek macierzystych w celu wyhodowania dużej ilości komórek, które niszczone są w chorobie HD. Komórki te mogą być również wytworzone tak, aby posiadały genetyczną wadę powodującą HD. Takie komórki mogą być następnie użyte podczas badań w celu odkrycia jak dokładnie działa choroba i jakie funkcje pełni prawidłowy gen w zdrowym mózgu.
- Opracowywanie nowych leków: ten sam rodzaj komórek macierzystych może być użyty do poszukiwania i testowania nowych leków.
- Zastępowanie utraconych komórek: w przyszłości naukowcy mają nadzieję na użycie komórek macierzystych w hodowli nowych, zdrowych komórek MSN, które będą mogły być przeszczepiane pacjentom, aby zastąpić komórki zniszczone przez chorobę.
Zrozumienie choroby Huntingtona i odkrywanie nowych leków
Jak wspomniano powyżej, gen huntingtiny zawiera fragment DNA zwany ‘powtórzeniami CAG’ – kawałek DNA zbudowany z trzech jednostek (C, A i G), które pojawiają się wielokrotnie w tej samej kolejności. Prawidłowy gen zawiera mniej niż 35 powtórzeń CAG, ale pacjenci chorzy na HD mają zmutowaną wersję, która posiada zbyt wiele powtórzeń CAG. Te dodatkowe kopie CAG tworzą wadliwą, toksyczną wersję białka huntingtiny. Im większa liczba powtórzeń CAG, tym bardziej toksyczne białko i prawdopodobnie tym wcześniej pojawiają się symptomy choroby.
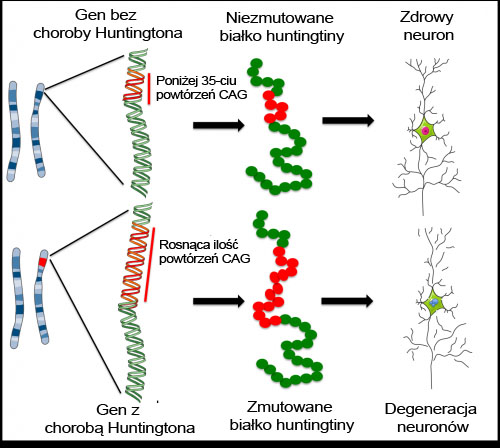
Naukowcy pracujący nad zmutowanym białkiem huntingtiny zastanawiają się – jak zmiana w tym jednym białku powoduje uszkodzenie komórek MSN? Niestety białko huntingtiny współpracuje z wieloma innymi białkami i wpływa na wiele różnych procesów w naszych komórkach, więc nie wiadomo dokładnie, w jaki sposób białko to uczestniczy w postępie choroby. Aby uzyskać odpowiedź na to pytanie, naukowcy używają różnych modeli choroby: systemów zaprojektowanych tak, aby reprezentowały chorobę HD w laboratorium i umożliwiały badanie choroby poza ciałem pacjenta. Komórki macierzyste używane są do rozwoju takich modeli w celu badania choroby i jej postępu.
Na przykład, indukowane pluripotencjalne (iPS) komórki mogę być stworzone z komórek mięśni lub skóry pacjentów z HD. Następnie mogę zostać namnożone w laboratorium przed przekształceniem ich w neurony. Dokonało tego konsorcjum naukowców pracujących nad HD składające się z sześciu amerykańskich i dwóch europejskich laboratoriów, a wyniki ich pracy zostały potwierdzone niezależnie przez laboratorium w Kalifornii. Komórki iPS, uzyskane od ludzi z różną liczbą powtórzeń CAG, mają taki sam kod genetyczny jak osoby cierpiące na HD. Kiedy te komórki iPS od różnych pacjentów zostaną użyte do wytworzenia komórek MSN, neurony z dłuższymi powtórzeniami CAG będą bardziej podatne na różne czynniki stresowe i wykazywać będą wiele z cech, jakie pojawiają się u chorych pacjentów. Naukowcy mają nadzieję, że ten model oparty na komórkach iPS pomoże im teraz w odkryciu jak dokładnie działa HD. Ta metoda, przynajmniej w teorii, może stanowić prawie nieograniczone źródło komórek MSN dotkniętych chorobą Huntingtona, ale ciągle potrzeba wiele pracy, aby stworzyć wydajny system do tworzenia komórek iPS z cechami choroby Huntingtona do przeprowadzania podobnych badań.
Możliwe jest użycie takiego modelu komórkowego do selekcjonowania potencjalnych leków, które mogą mieć zastosowanie terapeutyczne. Dzisiejsza technologia dostarcza mechanicznych systemów, które pozwalają naukowcom na testowanie milionów związków chemicznych w stosunkowo krótkim czasie w celu sprawdzenia czy mają one pożyteczny efekt dla komórek. Systemy te są bardzo przydatnymi narzędziami przy odkrywaniu leków, ale wymagają one ogromnych ilości komórek odpowiedniego typu (np. neuronów) z cechami choroby, która jest badana. Modele komórkowe oparte na iPS zawierające cechy HD mogą być używane do selekcjonowania leków, które następnie mogą być zdolne do modyfikowania choroby.
Zastępowanie utraconych komórek
Większość symptomów HD (lecz nie wszystkie) jest wynikiem utraty średnich neuronów kolczastych w mózgu. Do tej pory wysiłki naukowców skupiały się na otrzymaniu nowych komórek MSN w celu zastąpienia już zniszczonych. W roku 2000 i 2006 lekarze donieśli o przeszczepie neuronów płodowych do mózgów małej grupy pacjentów z HD. Osoby, u których przeszczep przetrwał, uzyskały poprawę symptomów zarówno ruchowych, jak i psychologicznych przez okres sześciu lat. To dostarczyło dowodów na poprawność założeń i oznacza, że przeszczep nowych komórek może spowodować poprawę symptomów HD, ale nie mówi nam jak niezawodna jest ta procedura. Po okresie sześciu lat symptomy HD znów się pogorszyły, co świadczy o tym, że proces nie jest jeszcze optymalny i potrzebnych jest więcej badań z użyciem komórek płodowych, aby opracować jak najlepsze wykonanie tej procedury. Możliwe jest, że udoskonalona procedura użycia tych płodowych komórek MSN mogłaby pomóc leczyć HD, a obecne próby kliniczne badają taką możliwość, ale istnieją też poważne naukowe i techniczne przeszkody. Na przykład użycie tkanki pobranej z usuniętego płodu dostarcza tylko ograniczonej ilości komórek, które nie mogą być oczyszczone czy udoskonalone. Komórki macierzyste mogłyby stanowić cenną alternatywę.
Naukowcy mają nadzieję na użycie embrionalnych komórek macierzystych lub komórek iPS jako nieograniczonego źródła średnich neuronów kolczastych do leczenia HD. Komórki iPS mogą być uzyskane od ludzi, którzy nie mają wady genetycznej w genie huntingtiny. Teoretycznie mogłyby one być też wytworzone ze skóry osób z HD, gdzie naukowcy poprawili genetyczną wadę używając techniki edytowania genów w taki sposób, aby komórki produkowały w pełni funkcjonalne białko huntingtiny. Komórki MSN są obecnie hodowane z genetycznie poprawnych komórek iPS i wykazują cechy zdrowych komórek: np. w laboratorium są tak samo odporne na śmierć komórkową jak komórki MSN od ludzi bez HD. Potrzebnych jest więcej badań, ale pewnego dnia ta metoda może oznaczać, że osoba chora na HD będzie mogła otrzymać przeszczep komórek mózgu z „poprawionych” komórek z jego własnej skóry.
Oba typy tych komórek (embrionalne komórki macierzyste i komórki iPS) mają zdolność do wytworzenia wszystkich komórek naszego ciała, więc wyzwaniem jest znalezienie drogi do pokierowania nimi tak, aby wytworzyły tylko komórki MSN. Obecnie wiele laboratoriów opracowało protokoły do różnicowania neuronów, które mają cechy komórek MSN z embrionalnych komórek macierzystych. Prowadzone badania testują jak blisko te neurony przypominają rzeczywiste komórki MSN i czy mogą one poprawić symptomy choroby po przeszczepieniu wykorzystując zwierzęcy model choroby Huntingtona. Pokłada się wielkie nadzieje na to, że takie komórki pewnego dnia będą nadawały się do przeszczepu ludziom z chorobą Huntingtona.
Zastosowanie komórek macierzystych w badaniach nad HD zostało stosunkowo niedawno wprowadzone. Chociaż komórki macierzyste już stały się przydatnymi narzędziami do badania postępu choroby HD i do poszukiwania nowych leków, potrzeba więcej pracy zanim leczenie z wykorzystaniem przeszczepu komórek macierzystych pacjentom będzie mogło być zastosowane. Główne pytania, które wymagają pilnej odpowiedzi to:
· Jak podobne są wyhodowane w laboratorium komórki MSN do tych znajdujących się w mózgu? Chociaż zostało pokazane, że mają pewne wspólne cechy, mogą nie być identyczne i należy to lepiej zrozumieć, aby metody do ich produkcji mogły być ulepszone.
· Jak długo komórki MSN z laboratorium mogą przetrwać po przeszczepieniu do mózgu i czy są zdolne do wykonywania swojej pracy?
· Czy komórki MSN z laboratorium byłyby zdolne do zintegrowania się z uszkodzonym systemem nerwowym dorosłego człowieka, ponownym połączeniem utraconych powiązań i zapewnieniem korzyści pacjentom?
· Czy przeszczep komórek MSN byłby bezpieczny dla pacjentów, szczególnie w dłuższej perspektywie?
Tak więc, komórki macierzyste nie mogą być obecnie używane do leczenia HD, ale badania nad nimi dostarczają praktycznych narzędzi dla naukowców zainteresowanych opracowaniem nowych metod w przyszłości. Praca ta jest kontynuowana w wielu międzynarodowych laboratoriach, a kilka z nich zaangażowanych jest w dwa konsorcja naukowe fundowane przez Komisję Europejską: Repair-HD i NeurostemcellRepair.
Broszura została opracowana przez Serafi Cambray i zrecenzowana przez Elena Cattaneo. Następnie broszura ta została zrecenzowana przez Anne Rosser.
Tłumaczenie na język polski: Mieszko Wilk.
Recenzja w języku polskim: Karolina Punovuori.
Zdjęcie główne przedstawiające neurony powstałe z embrionalnych komórek macierzystych wykonane przez Serafi Cambray. Średnie neurony kolczaste wybarwione na zielono przez białko zielonej fluorescencji wykonane przez Valentina Castiglioni. Ludzkie średnie neurony kolczaste wyhodowane z embrionalnych komórek macierzystych wykonane przez Charles Arber. Wszystkie inne zdjęcia i ryciny wykonane przez Serafi Cambray. Ryciny stworzone przy użyciu Servier Medical Art.
Ostatnia aktualizacja: